実験では捉えきれない原子分子レベルの情報を得ることができます。
第一原理計算シミュレーションに基づき、材料バルクや界面の構造および反応機構を詳細に解析し、実験分析の解釈の高精度化や材料設計指針の取得に貢献します。
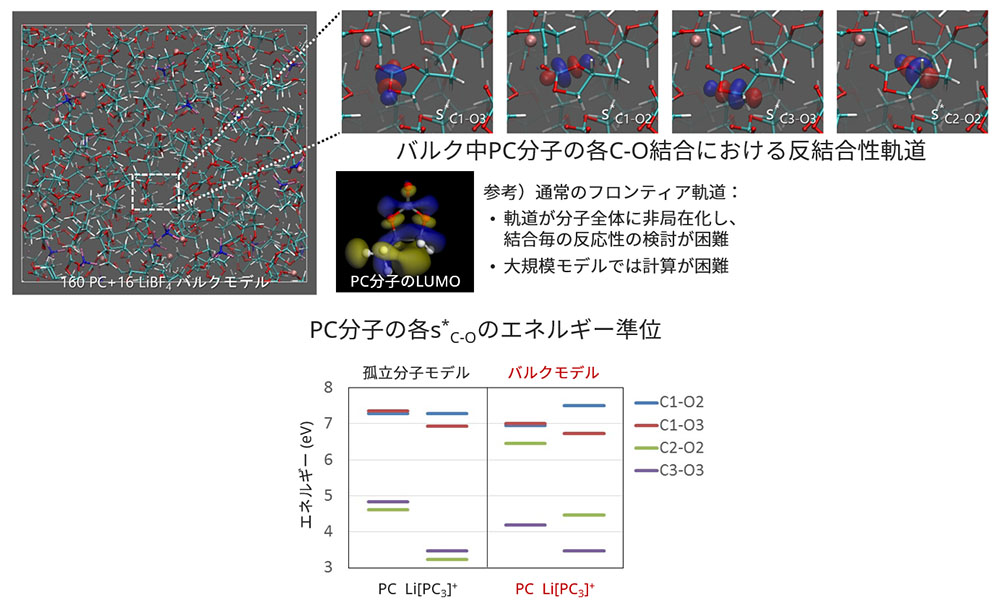
周囲の分子群の影響で、PC分子の反結合性軌道のエネルギー準位の序列が大きく変動することがわかりました。
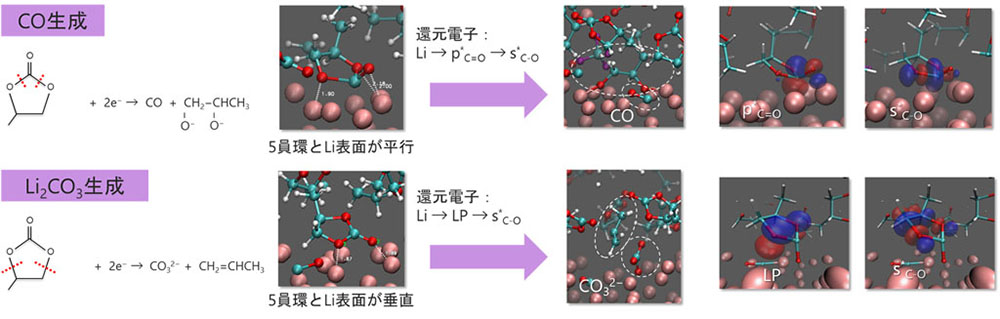
Li金属表面におけるPC分子の還元分解反応シミュレーションで確認された生成物は実験と対応しました。
表面吸着配向により還元電子の移動パスが変化し反応の多様性が発生することを解明しました。
掲載資料をダウンロードできます。
PDF形式
左のアイコンをクリックすると、別ウインドウで開きます。
資料のダウンロードにはお客様情報の入力が必要となります。